Research
The role of KRAS in the increased autophagic activity required for PDAC growth.
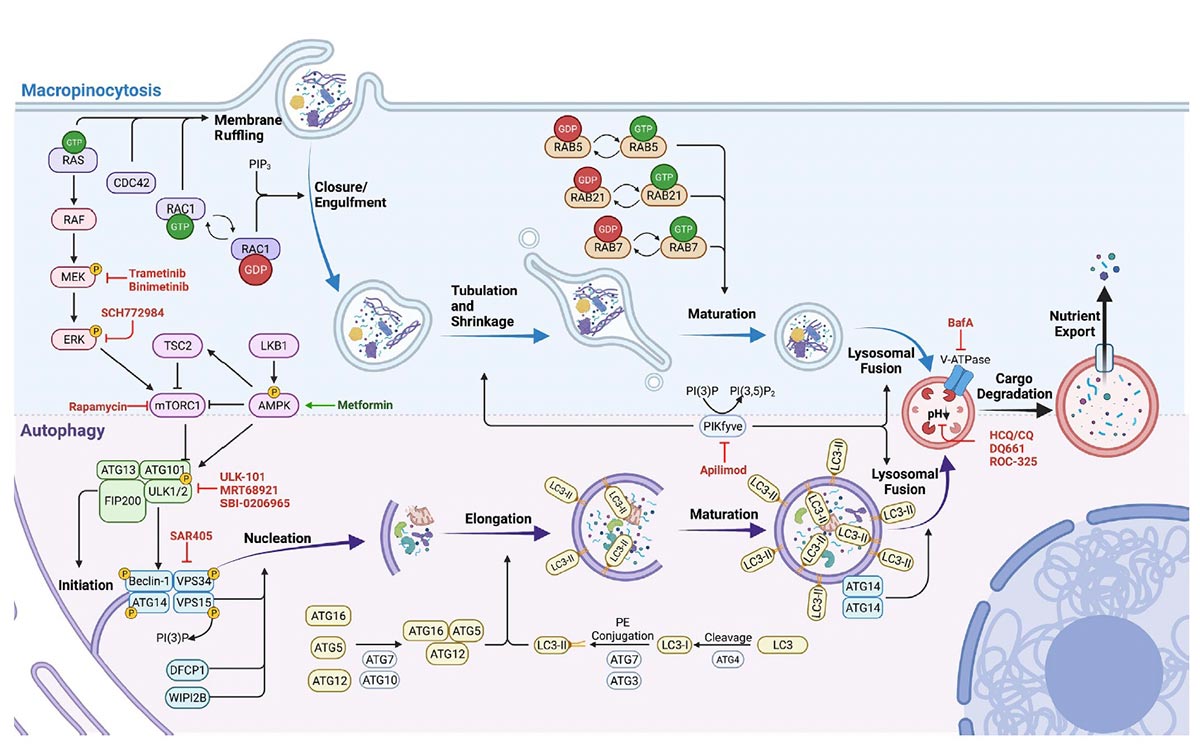
In a comprehensive mechanistic study (Bryant et al., Nature Medicine 2019), we found that the genetic suppression of KRAS increased autophagic flux, as did pharmacological inhibition of its effector, ERK MAPK. We speculated that ERK inhibition might enhance PDAC dependence on autophagy by impairing KRAS-driven metabolic processes. We utilized RNA-Seq data and metabolomics, among other techniques, to demonstrate that either KRAS suppression or ERK inhibition decreased both glycolytic and mitochondrial functions. Furthermore, we demonstrated that genetic or pharmacologic inhibition of autophagy synergistically enhanced ERK inhibitor-mediated anti-tumor activity. This finding led to the hypothesis that concurrent inhibition of ERK and autophagy may be an effective PDAC treatment. Based on this study, two clinical trials have been initiated by our group. One trial is being performed in collaboration with Dr. Shubham Pant at MD Anderson Cancer Center, and is an investigator sponsored clinical trial with Array BioPharma to study the combination of their MEK inhibitor binimetinib and the autophagy inhibitor hydroxychloroquine. Another trial is being performed in collaboration with Drs. Ashwin Somasundaram at UNC and Brian Wolpin at the Dana-Farber Cancer Institute to study the combination of the ERK inhibitor LY3214996 with hydroxychloroquine. We recently extended this study by demonstrating that upregulation of IGF receptor activity promote resistance to chloroquine treatment, and that cotargeting both ERK and IGF1R result in a greater dependence of PDAC cells on autophagic flux. Furthermore, IGF1R inhibition sensitized PDAC cells to ERK inhibitor and CQ treatment. Sensitization was enhanced when cells are cultured as three-dimensional spheroids as compared to two-dimensional culture and is particularly evident when cell toxicity, as opposed to proliferation, is used as an indicator (Stalnecker et al., Cancer Research, 2022).
Identification of novel signaling nodes for autophagy inhibition.
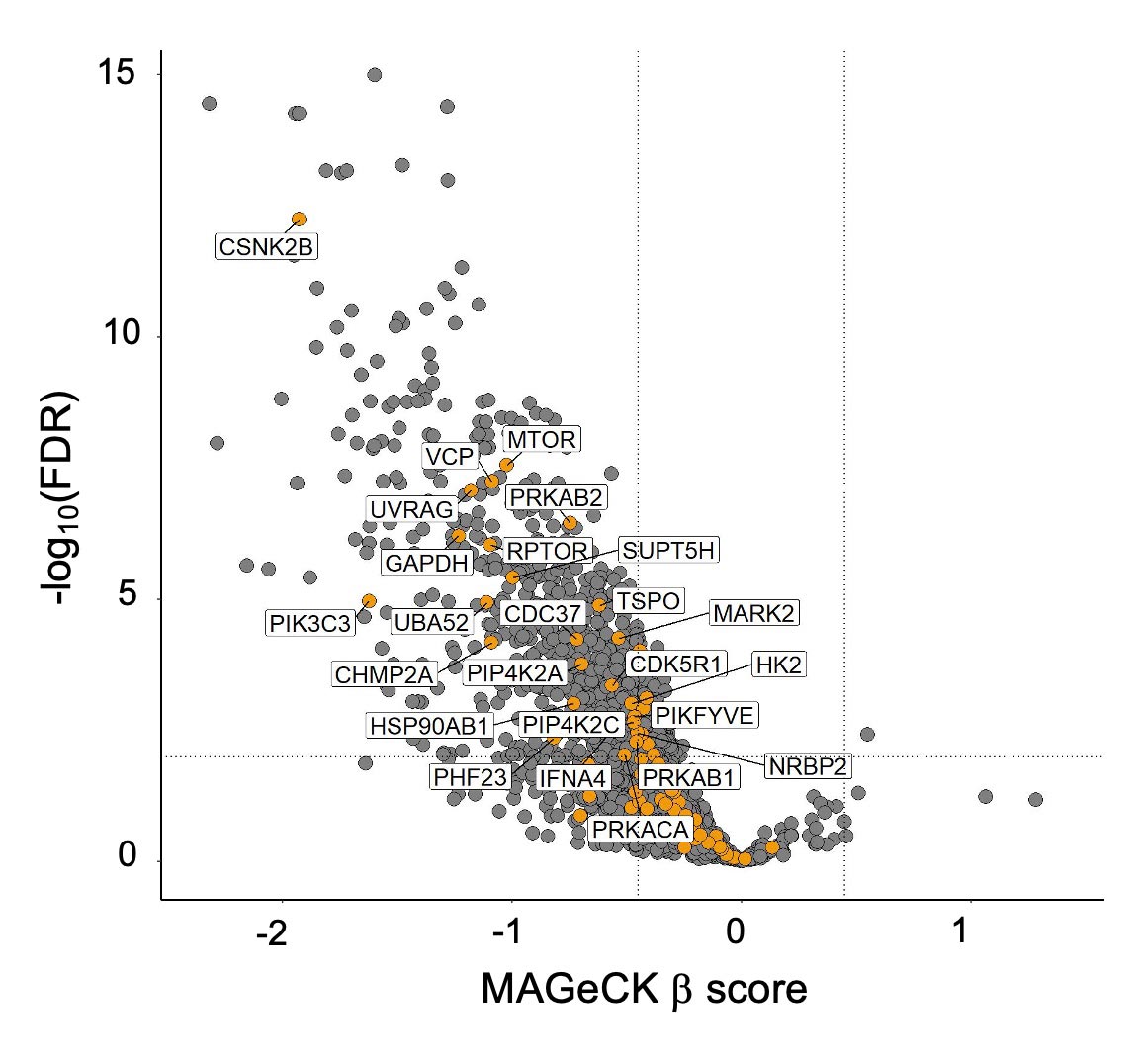
Hydroxychloroquine is limited by low potency and as a lysosomal inhibitor also disrupts non-autophagic processes. Ongoing projects in the laboratory are focused on determining unique ways to target the autophagy pathway for PDAC treatment. One target of particular interest is ULK, a kinase involved in the initiation step of the autophagic pathway. We are currently characterizing the effects of a panel of potent and selective ULK inhibitors on autophagic flux and PDAC proliferation alone and in combination with ERK MAPK inhibitors. We also aim to delineate compensatory mechanisms that arise following ULK inhibition. Additionally, we are pursuing the hypothesis that vertical inhibition of the autophagy pathway more completely inhibits autophagic flux and is a more efficacious strategy when paired with ERK inhibition. We are utilizing an autophagy-focused CRISPR library to address the contribution of each node in the autophagy pathway to autophagy-dependent cancer growth.
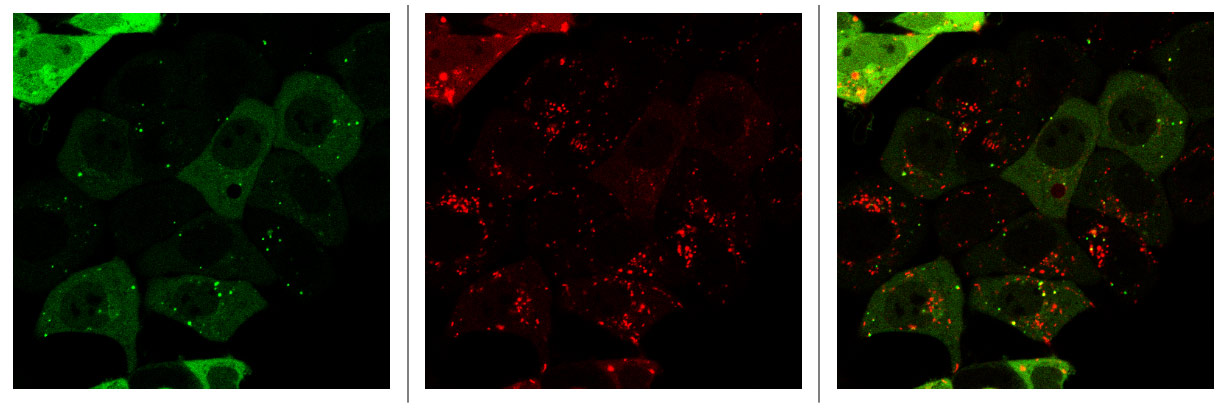
Elucidation of the interplay between autophagy and macropinocytosis in ERK MAPK inhibited pancreatic cancer cells.
We have shown that concurrent ERKi and CQ treatment synergistically enhances anti-tumor efficacy in KRAS-driven PDAC. While these preclinical results are promising, early clinical data has demonstrated that resistance to this treatment arises over time. KRAS or ERK MAPK depletion also results in downregulation of macropinocytosis in KRAS-driven PDAC. We hypothesize that autophagy induction, and macropinocytosis downregulation, via MAPKi can only be sustained transiently. The underlying mechanistic and signaling crosstalk between autophagy and macropinocytosis remain poorly understood. We hypothesize that there is a compensatory relationship between autophagy and macropinocytosis following blockade of the MAPK pathway and that this relationship influences the effect of autophagy inhibition in these models.
Determination of whether dual ERK and autophagy inhibition abrogates the growth of other RAS-driven cancers.
An important question is whether the combination of ERK MAPK and autophagy inhibitors will be effective in other KRAS-mutant cancers. For example, colorectal cancer (CRC) has a 45% frequency of KRAS mutations, but unlike PDAC, mutational loss of the APC tumor suppressor, not mutational activation of KRAS, is the initiating genetic event. Later mutations in KRAS, NRAS, or BRAF promote tumor progression, indicating distinct biological roles of RAS/MAPK in these cancer types. We are evaluating whether CRC is also responsive to combined ERKi and autophagy inhibition.